Enfermedades Raras
Enfermedad de Huntington

La enfermedad de Huntington es una enfermedad neurológica rara que pertenece al grupo de las demencias (disminución irreversible de la facultad mental) primarias y consiste en un trastorno hereditario, progresivamente degenerativo del sistema nervioso central (sistema formado por el encéfalo y la médula espinal), que se manifiesta en la vida adulta. Se caracteriza por corea (movimientos involuntarios del músculo), alteraciones del comportamiento y demencia.
La enfermedad de Huntington fue citada por primera vez en 1841, por Charles Waters de New York quien en una carta a un amigo describió una afección convulsiva singular, que era claramente hereditaria y más común en las clases bajas de la sociedad. Posteriormente en 1872 el médico estadounidense George Huntington describió una enfermedad, que ya su padre y su abuelo habían reconocido en algunos de sus pacientes, a la que llamó corea hereditaria. Se trataba de personas que tenían problemas al caminar o presentaban conductas inapropiadas; de aspecto casi cadavérico que se arqueaban y contorsionaban haciendo muecas, o el caso de dos hombres que acosaban constantemente a cualquier chica joven y no parecían ser conscientes de encontrar nada inapropiado en ello; los dos sufrían corea hasta el punto que les costaba mucho caminar y a la vista de cualquiera parecían estar drogados.
La comunicación de Huntington obtuvo una amplia difusión y aunque no se conocía cual era la causa, despertó bastante interés el seguimiento de las familias afectadas por la enfermedad. En 1916, C. P. Davenport y E. Muncie fueron capaces de clasificar a 962 enfermos de Nueva Inglaterra en cuatro familias que llegaron a Salem en el siglo XVII. P. R. Vessie en 1932, encontró en la investigación de una de esas familias, que la enfermedad provenía de tres hombres que llegaron a América en un barco, el John Withrop, en 1630 desde el pueblo de Bures en Inglaterra y que muchas mujeres descendientes de esta familia fueron quemadas en la hoguera durante el famoso juicio de Salem, al ser consideradas brujas. En los estudios genealógicos se encontró también que las familias afectadas solían tener gran cantidad de hijos, algunos con conductas criminales, especialmente crímenes sexuales, depresiones y suicidios.
La enfermedad de Huntington suele aparecer en la edad adulta, comienza a manifestarse entre los 30 y los 50 años, aunque la edad de comienzo puede ser muy variable, desde la infancia, hasta los 75 años. Afecta a 1 de cada 10.000 personas, siendo más frecuente en la raza blanca y sin predilección de sexo ni geográfica.
La causa de esta enfermedad hereditaria sigue sin conocerse, pese a haberse identificado el gen asociado a la misma. Dicho gen codifica una proteína, presente en la población, llamada hungtintina, que se encuentra en todas las neuronas del cerebro, pero de la que se desconoce su función. La destrucción neuronal en esta enfermedad parece estar ligada a la presencia de la mutación que provoca una hiperfunción de esta proteína que resulta tóxica y produce la apoptosis (muerte celular programada) neuronal.
Clínicamente pueden manifestarse en forma de deterioro físico, intelectual o emocional, aislados o combinados. El signo clínico más llamativo es la corea, palabra griega que significa danza, debido al movimiento característico de esta enfermedad. La corea comienza como una ligera inquietud motora que puede incluso pasar desapercibida para el paciente y sus familiares, progresa lentamente hasta llegar a ser incapacitante en el curso de unos quince o veinte años. Se producen sacudidas frecuentes irregulares y bruscas y movimientos de la cara, de las extremidades superiores o inferiores o el tronco. La marcha en la corea es dislocada y poco coordinada, como si el paciente fuera bailando, por lo que desde la antigüedad se ha llamado a la corea Mal de San Vito. Pueden presentar crisis convulsivas, más frecuentes y de pronóstico más severo en los pacientes más jóvenes. La gesticulación, la emisión de gruñidos y las dificultades para articular las palabras o tragar son muy llamativas y pueden orientar el diagnóstico.
La atención, la capacidad de juicio, la conciencia de la situación personal y la capacidad de tomar decisiones pueden alterarse desde las primeras etapas; por el contrario la afectación de la memoria sólo ocurre en las fases finales de la enfermedad.
Aparecen frecuentemente depresión, apatía (carencia de emociones) y aislamiento social, irritabilidad y desinhibición intermitente que excepcionalmente pueden ser los síntomas de comienzo de la enfermedad. En ocasiones los pacientes presentan delirios (trastorno de las facultades intelectuales, que se manifiesta por una serie de pensamientos erróneos, disparatados e inaccesibles a toda crítica, que puede o no acompañarse de alteraciones de la conciencia) y comportamientos obsesivo-compulsivos.
El diagnostico de sospecha de la enfermedad de Huntington se basa en los hallazgos clínicos: movimientos coreiformes y demencia asociados con historia familiar positiva.
El escáner y la resonancia magnética nuclear cerebral suelen poner de manifiesto en las fases medias y tardías de la enfermedad la atrofia (disminución de volumen y peso de un órgano) de núcleo caudado y en las fases finales una atrofia cerebral difusa y severa.
Anatomopatológicamente la enfermedad afecta predominantemente al núcleo estriado, existiendo gliosis (proliferación de la red neurológica) y pérdida neuronal sobre todo a nivel de núcleo caudado y putamen, pero no se encuentran placas ni ovillos como en la enfermedad de Alzheimer.
Desde el punto de vista neuroquímico existe una disminución de GABA y un aumento de la concentración de lactato en todos los ganglios basales y también se aprecia la disminución de otros neurotransmisores (sustancia liberada por las terminaciones nerviosas bajo el influjo de una excitación, transmitiendo la información de una neurona a otra) como la sustancia P y las encefalinas.
El diagnóstico de confirmación se realiza mediante técnicas de genética molecular.
El diagnóstico diferencial debe hacerse con esquizofrenia, corea familiar benigno, ataxias hereditarias, acantocitosis neural, enfermedad de Alzheimer, enfermedad de Pick o enfermedad de Creutzfeldt Jakob.
Los hijos de pacientes asintomáticos con riesgo de padecer corea de Huntington deben recibir consejo genético adecuado, ya que un resultado positivo puede tener graves consecuencias tanto emocionales como sociales.
La evolución es lentamente progresiva, llevando a la muerte normalmente al cabo de 15 a 25 años. Inicialmente el paciente está normal hasta que sobreviene la llamada fase temprana intermedia; en la cual comienza a manifestarse el proceso degenerativo, produciéndose el deterioro funcional progresivo y la pérdida de habilidades, requiriendo, en las últimas fases, ayuda total para cualquier actividad de la vida diaria. Los pacientes alcanzan un estado de postración y debilidad extremas y fallecen a consecuencia de complicaciones, por lo general infecciones sobre todo respiratorias.
En la actualidad no existe tratamiento curativo o que pueda detener el avance de la enfermedad. Existen algunos medicamentos útiles parcialmente para el control de ciertos síntomas como movimientos coreiformes, depresión, apatía e irritabilidad; estos fármacos son: fenotiazinas, haloperidol, benzodiazepinas, olanzapina e inhibidores de la recaptación de serotonina para la depresión; no obstante su efecto tiende a ser menor según avanza la edad del paciente.
Es fundamental tener en cuenta para todas aquellas personas que se vayan a encargar del cuidado de enfermos con la enfermedad de Huntington, una serie de consideraciones que resultan de ayuda:
1.- Vigilar el riesgo de atragantamiento, debido a la dificultad para la deglución y a que muchos enfermos tienden a comer muy deprisa y a meterse demasiada comida en la boca, se aconseja que coman en un ambiente tranquilo y sin ningún tipo de distracción.
2.- Prevenir las caídas, ya que la marcha es muy tambaleante y no puede mantener el equilibrio. Son frecuentes durante el traslado del enfermo, especialmente al acostarlo y levantarlo.
3.- Extremar la paciencia, estos enfermos no pueden controlar los impulsos, cuando quieren algo, lo quieren inmediatamente y no obtenerlo les genera más ansiedad e irritabilidad. Presentan además una marcada tendencia inevitable a la repetición, el paciente entiende lo que usted le contesta pero tiene dificultad para cambiar de tema pues se le ha fijado en el cerebro.
4.- Adecuar la ingesta calórica, tienden a perder peso especialmente ante situaciones de estrés o durante el período de adaptación a un nuevo entorno, cambio de domicilio, ingreso en residencias, o cambio de cuidadores.
5.- Conocer su falta de expresión, ya que la afectación muscular, especialmente de la cara hace que el enfermo presente una expresión de aburrimiento e indiferencia. La dificultad para sonreír mientras escucha o habla induce a creer que está triste o desinteresado. Otros cambios posturales inducen a pensar que el paciente está incómodo en presencia del cuidador, por lo que éste tiende erróneamente a comunicarse cada vez menos con el enfermo.
6.- Establecer rutinas ya que esto les facilita coordinar los pensamientos y procesar la información. Cuando vaya a ocurrir algo fuera de la rutina diaria, se les debe avisar con antelación y reiteradamente a fin de evitarle sorpresas.
7.- Si el paciente es fumador, no debe impedírsele fumar pues adquiere una importancia vital ya que ante tantas pérdidas, trabajo, amigos, habilidades, autosufiencia etc. en muchas ocasiones representa para él lo único que le queda. Se deben establecer rutinas en cuanto a horarios, sitios donde no se debe fumar y vigilancia para evitar quemaduras y otros accidentes.
La enfermedad de Huntington se hereda como un rasgo genético autosómico dominante, aunque también se han descrito casos esporádicos. El gen responsable de la enfermedad, conocido como IT15, se ha localizado en el brazo corto del cromosoma 4 (4p16.3).
Otras notas
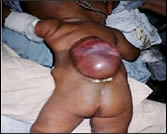
La espina bífida es una malformación congénita del tubo neural, que se caracteriza porque uno o varios arcos vertebrales posteriores no han fusionado correctamente durante la gestación y la médula espinal queda sin protección ósea.

La sociedad nos ha bautizado "discapacitados". Sin embargo, nosotros no solemos identificarnos con esta denominación. En general, la asociamos a la falta de integración que nos reserva el medio. Muchas veces nos enojamos cuando nos sentimos rechazados...

La Policía de Seguridad Aeroportuaria (PSA) aprobó, por Disposición 752/2012, el 'Protocolo General de Actuación Ante Personas con Discapacidad Nº 1', que será de aplicación obligatoria para todo el personal policial de las terminales aéreas.